
Table Of Contents
Intro to LaJolla
My PhD thesis was about understanding biomolecules, and one part of that is finding structural similarities between them (full thesis PDF). That work became LaJolla - a 3D alignment algorithm for RNA and proteins, originally published in Algorithms in 2009.
LaJolla lets you find similar protein structures - or subparts of them, like binding sites - and, more novel at the time, similar RNA structures. With the rise of mRNA that turned out to be a genuinely useful capability. RNA does far more than shuttle information between DNA and proteins: it folds into specific 3D shapes, and those shapes have function. Being able to search for similar RNA folds matters.
LaJolla uses an efficient index to find similar structures really really fast. Which is important if you want to screen hundreds of thousands of biomolecues for scientifically relevant similarities.
The original code was last touched in 2013 and then lay dormant. At some point I moved it off SourceForge and onto GitHub (raphaelbauer/lajolla), where it had been quietly waiting for someone to care about it again.
The idea behind LaJolla was simple to state and fiddly to get right:
- Translate a 3D biomolecule into a 1D string.
- Index that string.
- Run fast searches against the index to find promising candidates.
- Only then superimpose the good-looking candidates with a potentially costly geometric algorithm.
The secret sauce was step one: how do you turn a 3D structure into a linear string without throwing away the shape? And a nice property falls out of the design - it works for any molecule organized as a chain. Proteins are chains. RNA is a chain. The same machinery handles both.
The basic idea, and how it actually works
The expensive part of structural comparison is the geometry: rotating and translating one molecule onto another in 3D space and measuring how well they overlap. Doing that for every candidate in a large database is hopeless. So LaJolla does what search engines do with text - it builds an index and only pays the expensive cost for the handful of candidates that look worth it.
The trick that makes this work for molecules is a structural alphabet.
A protein backbone can be described almost completely by two angles per residue: the phi and psi dihedral angles. These are the two main rotatable bonds along the backbone, and together they capture the local shape of the chain - whether a stretch is an alpha helix, a beta strand, a turn, and so on. If you walk down the backbone and read off (phi, psi) at every residue, you have a compact numerical fingerprint of the fold.
LaJolla takes those continuous angles and discretizes them into letters. Each residue’s local geometry becomes one character. A whole protein becomes a string over a small alphabet - conceptually the same move as writing down a protein’s amino-acid sequence, except these letters encode shape, not chemistry.
Once a structure is a string, everything gets easier:
- Indexing. LaJolla slides a window along the string and indexes the resulting n-grams (short overlapping substrings of shape-letters). This is the
SequenceDB- an in-memory index from n-gram to the structures that contain it. - Fast candidate search. To find structures similar to a query, you turn the query into its own shape-string, look up its n-grams, and collect the structures that share many of them. This is cheap - it is string matching, not geometry.
- Expensive refinement, only where it counts. For each promising candidate, LaJolla superimposes the matched fragments in real 3D space using a singular value decomposition (SVD) to find the optimal rotation and translation, then scores the fit with the TM-score, a standard length-normalized measure of structural similarity where 1.0 means identical and low values mean unrelated.
RNA gets the same treatment with different angles. Instead of phi/psi, LaJolla uses the RNA backbone’s eta/theta pseudo-torsion angles - which describe RNA conformation much as phi/psi describe proteins - and alternatively the suite codes from the Richardson/Pyle RNA conformer work, a ready-made discrete vocabulary for RNA backbone geometry. Same pipeline, different alphabet.
So the whole design is: shape becomes text, text becomes an index, the index gives you candidates, and geometry is spent only on candidates that already look right. That is what let it search structure databases quickly back in 2008-2013.
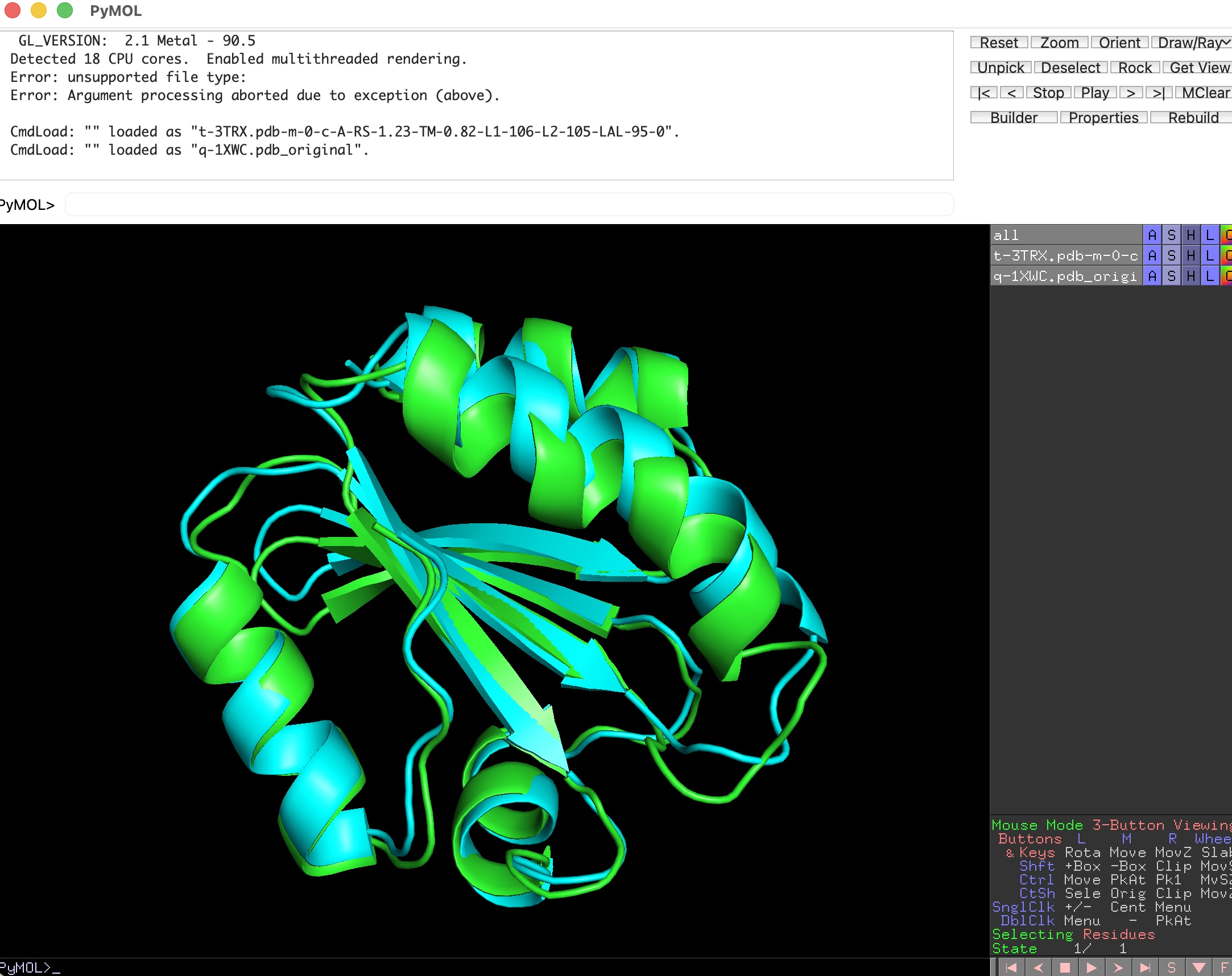
It was good to see PyMol still alive and kicking! The expensive last step, visualized by PyMol: two structures superimposed in 3D after LaJolla found them as candidates and computed the optimal rotation and translation. This is the geometry you only want to pay for on candidates that already look promising.
Modernizing LaJolla with Claude
The starting point: LaJolla’s source, essentially untouched for about 13 years. It still worked - but everything around it had aged.
Here was the problem. The code worked, but the world around it had moved on more than a decade:
- BioJava was ancient. LaJolla leans on BioJava to read structure files and to do the 3D superposition. The version it used (3.0.7) is long gone. The package names changed (
org.biojava.bio.structurebecameorg.biojava.nbio.structure), APIs were removed, and the old library was served from a dead Maven repository that no longer exists. - Java 8, old Maven, old everything. The build itself was from another era. Basically every dependency was outdated.
None of this was hard in the sense of being intellectually difficult. It was hard in the sense of being tedious - hundreds of tiny breakages, each requiring you to go and learn what the new API looks like, what got renamed, what got removed, what subtly changed behavior. The honest calculation for a dormant side project was: modernizing it is clearly possible, but the time cost is not worth it.
But we now have Claude.
So I pointed Claude Code at the repository and asked it to modernize LaJolla. It went remarkably smoothly. The migration touched the build (Java 8 to Java 25, Maven refreshed, all dependencies pulled from Maven Central), the BioJava 3 to BioJava 7 API move, the CLI entry points, the logging stack, and the test suite (moved to JUnit 5).
The single most valuable thing I had going in was tests. LaJolla came with a large suite of regression tests built around real structure files - including the crucial invariant that aligning a structure against itself must produce a perfect result (RMSD 0.0, TM-score 1.0). That suite is the behavior oracle. It is what let me trust that the modernized code still computes the same answers as the 2013 code, rather than something that merely compiles and looks plausible.
That trust matters, because the migration did surface a genuine subtlety. When BioJava’s old SVDSuperimposer was replaced with the newer SuperPositionSVD, the rotation matrix has to be transposed to match the row-vector convention BioJava’s Calc.rotate expects. Get it wrong and superpositions are subtly, silently incorrect. The tests caught exactly this - a good reminder that “it builds” is not the same as “it’s right,” and that a well-tested legacy project is far safer to hand to an AI than an untested one.
Then: hundreds of machines. Now: five seconds.
There is a second kind of aging worth talking about, and it is not the code - it is the compute around it.
Back when LaJolla was active research, screening large structure databases was not something you did on a laptop. The heavy all-against-all comparison runs went onto the cluster at the Zuse Institute Berlin (ZIB) - hundreds of machines chewing through hundreds of thousands of structures in parallel. That was simply what “large scale” meant in 2008-2013. The whole architecture of LaJolla - index first, spend geometry only on the survivors - was in part a response to that reality: compute was precious, so you did everything you could to avoid the expensive step.
Here is the anecdote that made me stop and blink. In 2009, running LaJolla’s own test suite took me several minutes. Today, on an M5 Mac, the entire suite - the same regression tests over the same real structure files - runs in about five seconds.
Same algorithm. Same tests. Minutes to seconds, on a machine that fits in a backpack.
It makes me genuinely wonder how much of the original distributed, cluster-bound machinery you would even need today. A lot of what once demanded a supercomputer center now fits comfortably on a single modern workstation. The problem did not get smaller - the hardware got absurdly better.
LaJolla as a scientific project - did it stand the test of time?
Here is the part I was genuinely unsure about. I left academia a long time ago. I have no current view of where structural search actually stands in 2026. So I asked Claude to evaluate whether LaJolla’s basic idea had aged well, and to tell me what it would be measured against today.
The short version it gave me:
If you were writing LaJolla today, Foldseek is the tool it would be measured against - it’s the same structural-alphabet-plus-fast-search idea taken to production scale with a learned alphabet. For accuracy benchmarking you’d compare against US-align/TM-align, and for RNA against US-align and the suite-based RNA aligners. LaJolla’s design choices (structural alphabet, n-gram indexing, TM-score) have aged well - they’re exactly the ideas that won.
Digging into that a little:
- Foldseek (2022 onward) is the breakout modern tool for fast structure search. Its architecture is strikingly familiar: it turns each residue’s local structural environment into a letter from a learned 3Di alphabet, then runs fast sequence-style search over those letters and only refines the good hits geometrically. That is the exact shape of LaJolla’s pipeline - shape becomes text, text is searched fast, geometry is spent only on candidates. The main advance is that Foldseek’s alphabet is learned from data and describes each residue’s spatial neighbors, rather than being a hand-designed discretization of backbone angles. The paper reports it running four to five orders of magnitude faster than Dali, TM-align and CE at comparable sensitivity - which is what made searching the ~200M+ structures of the AlphaFold era actually feasible.
- TM-align / US-align remain the accuracy reference standard - the slower, careful aligners you benchmark against. LaJolla already scores with the TM-score, so it was speaking the right language all along.
- For RNA, US-align and the suite-based RNA aligners are the modern references - and they draw on the same RNA backbone conformer vocabulary that LaJolla used for its RNA alphabet.
It is genuinely nice to see that the field converged on the same core intuition: discretize local 3D shape into a string, search the strings fast, and only pay for geometry on the survivors. LaJolla bet on that in the late 2000s and the bet held up.
It is less nice - and I will be honest about this - to see that LaJolla was not referenced in the Foldseek paper. That is how research often goes: good ideas get reinvented independently, and the version that arrives at the right moment, with the right engineering and the right dataset behind it, is the one that gets remembered. No hard feelings. It is a real compliment to be, in hindsight, on the right track.
Where next?
Bringing LaJolla back was supposed to be a nostalgia project. It turned into something more tempting.
A few directions I am genuinely toying with:
- Beat Foldseek. Partly for the sport of it - and yes, partly because LaJolla was not in their references and it would be fun to close that loop. Not out of spite; it is just an honest benchmark worth chasing. The two approaches overlap enough that a serious head-to-head would be interesting regardless of who comes out ahead.
- Improve the algorithm itself. The structural alphabet, the indexing, the scoring - all of it predates a decade of ideas, including learned alphabets like Foldseek’s 3Di. There is real room to modernize the science, not just the build.
- Speed and distribution. Now that a single machine does what a cluster used to, the interesting question is how far a lean, well-engineered implementation can go. I have even been tempted to rewrite the core in Go - simple, dependency-free static executables you can drop on any machine and run, no JVM required.
None of this is committed. But it is the first time in over a decade that touching LaJolla feels like the start of something rather than a museum visit.
Summary
LaJolla was a 3D structural alignment tool for proteins and RNA, built on one durable idea: translate a molecule’s shape into a string, index it, search fast, and reserve expensive geometry for the promising candidates.
Two things stand out from bringing it back to life:
- Modernization that used to be uneconomical is now cheap. A decade-dormant project with an ancient dependency stack went from “not worth the tedium” to a smooth afternoon’s work with an AI agent doing the migration. The key enabler on my side was a strong regression test suite - that is what turned “it compiles” into “it still computes the same answers,” and it is what caught a subtle superposition bug during the migration.
- The core idea aged well. The modern state of the art, Foldseek, works in essentially the same way LaJolla did - a structural alphabet plus fast string search plus geometric refinement - just at a scale and with a learned alphabet that the 2010s could not have delivered. The design choices held up.
Sometimes the most satisfying thing you can learn about old work is that the field eventually agreed with it.
References
- LaJolla (original paper). Bauer, R.A., Rother, K., Moor, P., Reinert, K., Steinke, T., Bujnicki, J.M., Preissner, R. “Fast Structural Alignment of Biomolecules Using a Hash Table, N-Grams and String Descriptors.” Algorithms 2, no. 2 (2009): 692-709. mdpi.com/1999-4893/2/2/692
- LaJolla source code. github.com/raphaelbauer/lajolla
- PhD thesis. refubium.fu-berlin.de (PDF)
- Foldseek. van Kempen, M., Kim, S.S., Tumescheit, C., et al. “Fast and accurate protein structure search with Foldseek.” Nature Biotechnology 42 (2024): 243-246. nature.com/articles/s41587-023-01773-0
- TM-align / TM-score. Zhang, Y., Skolnick, J. “TM-align: a protein structure alignment algorithm based on the TM-score.” Nucleic Acids Research 33, no. 7 (2005): 2302-2309. academic.oup.com/nar/article/33/7/2302/2401364
- US-align. Zhang, C., Shine, M., Pyle, A.M., Zhang, Y. “US-align: universal structure alignments of proteins, nucleic acids, and macromolecular complexes.” Nature Methods 19 (2022): 1109-1115. nature.com/articles/s41592-022-01585-1
- RNA eta/theta pseudo-torsions. Wadley, L.M., Keating, K.S., Duarte, C.M., Pyle, A.M. “Evaluating and Learning from RNA Pseudotorsional Space: Quantitative Validation of a Reduced Representation for RNA Structure.” Journal of Molecular Biology 372, no. 4 (2007): 942-957. pmc.ncbi.nlm.nih.gov/articles/PMC2720064
- RNA suite codes. Richardson, J.S., Schneider, B., Murray, L.W., et al. “RNA backbone: consensus all-angle conformers and modular string nomenclature (an RNA Ontology Consortium contribution).” RNA 14, no. 3 (2008): 465-481. rnajournal.cshlp.org/content/14/3/465